Abstract
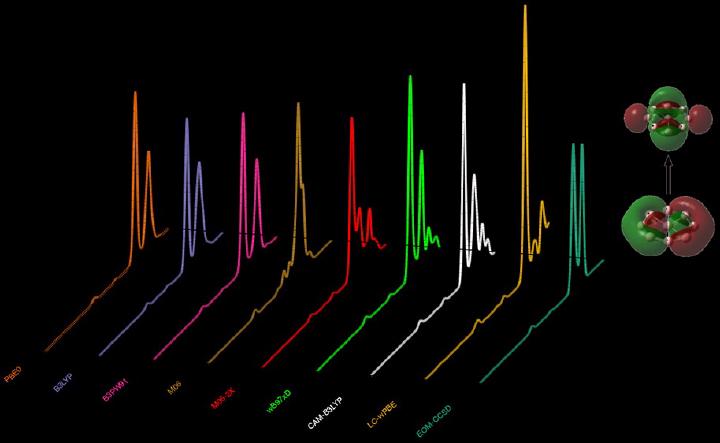
We benchmark various quantum chemical methods for calculating the optical absorption in planar boron wheel clusters. The geometries of neutral planar boron wheels B7, BB8, and BB9 clusters are optimized at the coupled-cluster singles doubles level of theory. The optical absorption spectra of these clusters are calculated using three wave-function-based methods, namely, configuration interaction singles, random phase approximation, and equation-of-motion coupled-cluster singles doubles (EOM-CCSD) as well as using a time-dependent density-functional-theory-based method using various hybrid and long-range-corrected exchange and correlation functionals. There is an ample variation in the optical absorption spectra computed using different density functionals. When compared to the EOM-CCSD spectrum, an excellent agreement is provided by CAM-B3LYP functional, followed by ωB97xD functional. PBE0, B3LYP, and B3PW91 functionals agree among each other. However, their spectra are red-shifted with respect to the EOM-CCSD counterpart. On the basis of the natural transition orbital analysis, the nature of optical excitation is also discussed.
Search content by categories
Ravindra Shinde
Research Scientist
I am a theoretical and computational condensed matter physicist and quantum chemist. I am currently working as a researcher at the University of Twente, the Netherlands. I am also the founder of The Science Dev.